Study reveals mechanism used to regulate chaperone proteins, which guide other proteins into proper shapes
Newswise — DALLAS – Feb. 11, 2021 – A study led by UT Southwestern has identified a mechanism that controls the activity of proteins known as chaperones, which guide proteins to fold into the right shapes. The findings, published online today in Nature Communications, could shed light on hundreds of degenerative and neurodegenerative diseases caused by protein misfolding, such as Alzheimer’s, Parkinson’s, and Huntington’s, potentially leading to new treatments for these devastating conditions.
Every protein in the body is originally produced in a linear chain, with amino acid building blocks strung together one after another. But to fulfill their roles in cells, explains study leader Lukasz Joachimiak, Ph.D., assistant professor in the Center for Alzheimer’s and Neurodegenerative Diseases at UT Southwestern, these chains need to fold into precise shapes. Chaperones help proteins accomplish this by protecting their vulnerable portions while they shift into position and steering them to adopt the proper shape.
Every cell has a variety of chaperones that recognize and act on individual protein types. However, every chaperone isn’t active all the time, Joachimiak says. Unknown regulatory mechanisms appear to control when certain chaperones step in to guide their respective proteins to fold and when they stand aside.
Joachimiak, also a member of the Peter O’Donnell Jr. Brain Institute, and his colleagues studied a family of chaperone proteins known as Hsp40s that work in combination with other chaperones known as Hsp70s. Members of these co-chaperones are involved in the proper folding of many proteins, including tau, which play a key role in causing Alzheimer’s disease when it’s misfolded.
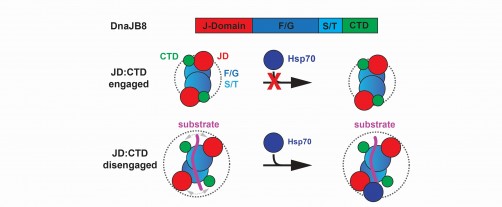
Hsp40 chaperones bind to Hsp70s through a specific portion on the Hsp40s called the J domain. But how the Hsp40s turn off this binding when it is not needed has been unclear.
To help answer this question, Joachimiak and his colleagues used a specific Hsp40 called DnaJB8 as a model. When the researchers genetically modified these proteins to glow green inside cells, they found that they didn’t just exist as individual, free-floating units – the DnaJB8 chaperones tended to form aggregates, suggesting they had some way to stick to each other. They retained this ability to agglomerate when they were isolated in petri dishes.
Using computer modeling and guided by biochemical experiments, the researchers discovered that two separate parts of this chaperone were drawn to each other through a type of chemistry called electrostatic interactions: Part of the J domain was drawn to a different part of this protein called the C-terminal domain through charged interactions. Modeling also showed that the J domain and the C-terminal domain stuck together on single molecules as well.
Joachimiak and his team validated these findings on real DnaJB8 proteins using a technique called solid-state nuclear magnetic resonance. They also showed that the J and C-terminal domains stuck to each other when they were isolated from the full DnaJB8 molecule.
The researchers suspected that the interaction between these two domains could prevent DnaJB8 from binding to its co-chaperone, an Hsp70, preventing them from jointly doing their job of guiding protein folding. Sure enough, experiments showed that the C-terminal domain of DnaJB8 competed with an Hsp70 called HspA1A when it was added to DnaJB8 in a test tube, blocking HspA1A from binding to the J domain when the C-terminal domain was bound instead.
Joachimiak notes that something may go awry in this or other regulatory mechanisms that control the activity of chaperones in protein misfolding diseases. Finding ways to control this activity through pharmaceuticals or other means could provide a new way to treat these conditions to attack the problem at its source.
“We may be able to leverage this mechanism to directly target these chaperones, activating them at will,” says Joachimiak, who is also an assistant professor of biochemistry and an Effie Marie Cain Scholar in Medical Research. “Our results could have an impact on hundreds of diseases where proteins become bad players by misfolding.”
Other UT Southwestern researchers who contributed to this study include Bryan D. Ryder, Sofia Bali, and Jaime Vaquer-Alicea.
This work was supported by a grant from The Welch Foundation (I-1928-20170325).
About UT Southwestern Medical Center
UT Southwestern, one of the premier academic medical centers in the nation, integrates pioneering biomedical research with exceptional clinical care and education. The institution’s faculty has received six Nobel Prizes, and includes 23 members of the National Academy of Sciences, 17 members of the National Academy of Medicine, and 13 Howard Hughes Medical Institute Investigators. The full-time faculty of more than 2,500 is responsible for groundbreaking medical advances and is committed to translating science-driven research quickly to new clinical treatments. UT Southwestern physicians provide care in about 80 specialties to more than 105,000 hospitalized patients, nearly 370,000 emergency room cases, and oversee approximately 3 million outpatient visits a year.

